What Is the ANDA Process?
The ANDA process is the legal pathway the U.S. Food and Drug Administration (FDA) uses to approve generic drugs. It’s not a shortcut-it’s a carefully designed system that lets companies sell cheaper versions of brand-name medications without repeating expensive clinical trials. The goal? Get safe, effective generics to patients faster and at lower cost. Since the 1984 Hatch-Waxman Act created this system, generic drugs have gone from covering 19% of prescriptions to over 90% today. That’s not luck-it’s regulation working as intended.
Why the ANDA Process Exists
Before 1984, generic drug makers had to prove safety and effectiveness from scratch, just like brand-name companies. That meant spending hundreds of millions and waiting over a decade. The result? Few generics entered the market, and prices stayed high. The Hatch-Waxman Act changed that. It gave innovator companies a limited time of exclusivity to recoup R&D costs, while creating a clear path for generics to follow once patents expired. The law balanced two goals: protecting innovation and promoting competition. Today, that balance saves U.S. consumers an estimated $2.2 trillion over the last decade.
Core Legal Requirements for ANDA Approval
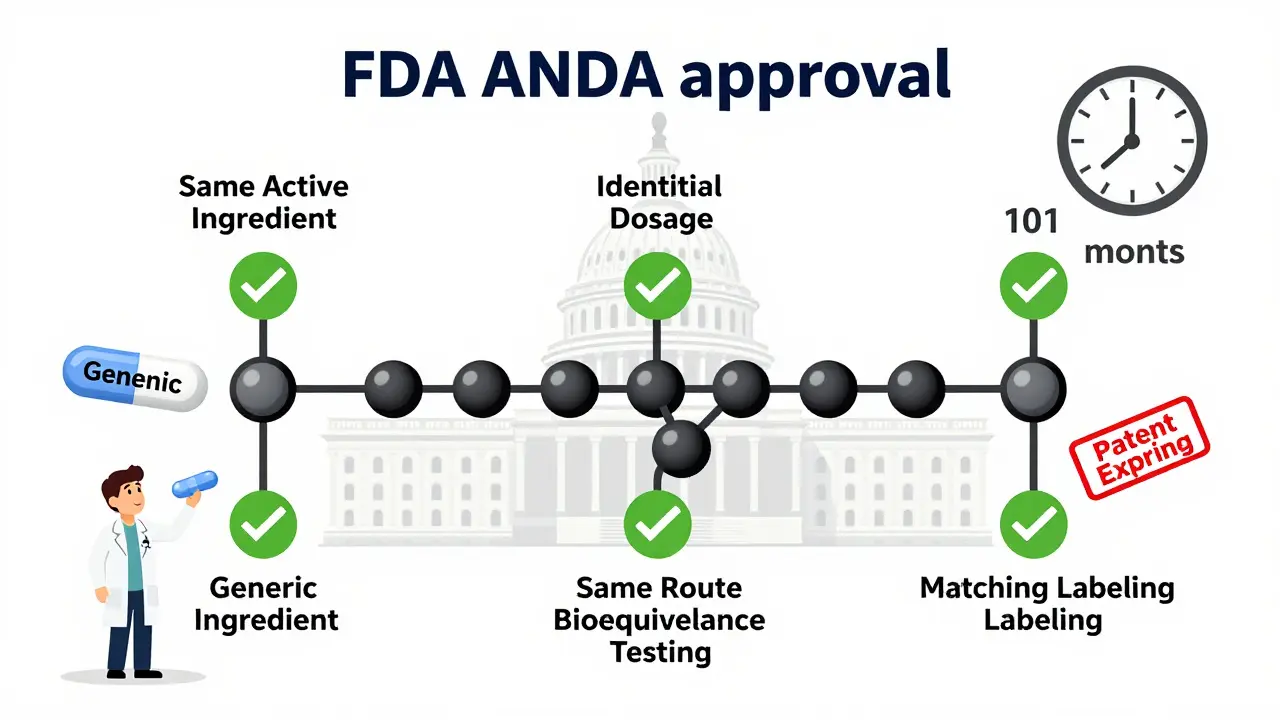
To get an ANDA approved, a generic drug must meet five strict legal and scientific standards:
- Same active ingredient: The generic must contain the exact same active pharmaceutical ingredient (API) as the brand-name drug. No substitutions. No alternatives. Even minor changes require a separate application.
- Identical dosage form and strength: If the brand drug is a 10mg tablet taken orally, the generic must be a 10mg tablet taken orally. Changing the form-say, to a capsule or liquid-requires a different approval pathway.
- Same route of administration: Whether it’s injected, swallowed, inhaled, or applied topically, the generic must match the brand’s method of delivery.
- Bioequivalence: This is the most critical technical requirement. The generic must deliver the same amount of drug into the bloodstream at the same rate as the brand drug. The FDA requires proof that the 90% confidence interval for peak concentration (Cmax) and total exposure (AUC) falls between 80% and 125% of the brand drug’s values. This isn’t theoretical-it’s tested in real people using controlled clinical studies.
- Same labeling: The generic’s prescribing information must match the brand’s, except for the manufacturer’s name and certain minor differences tied to the generic status. No extra claims. No hidden warnings.
What the FDA Expects in the Application
An ANDA isn’t just a form. It’s a massive technical dossier. The FDA requires submissions in eCTD format, which organizes data into 15 modules. Key components include:
- Chemistry, Manufacturing, and Controls (CMC): Detailed descriptions of how the drug is made, tested, and packaged. This includes raw material sourcing, equipment specs, process validation, and stability data showing the drug won’t degrade over time.
- Bioequivalence study reports: Must follow FDA guidance and include full protocols, statistical analysis, and subject demographics. The FDA has rejected applications because the study didn’t include enough participants or used the wrong fasting state.
- Patient labeling: Must be identical to the brand’s in content, even if formatting differs slightly.
- Patent certifications: Applicants must certify one of four things about the brand’s patents: (I) no patent listed, (II) patent expired, (III) patent will expire on a specific date, or (IV) the patent is invalid or won’t be infringed. Choosing Paragraph IV triggers potential lawsuits from the brand company.
- Form FDA-356h and FDA-3674: Official application and fee forms. Missing these means the FDA won’t even review your application.
Costs and Fees
While the ANDA process is far cheaper than developing a new drug, it’s still expensive. For fiscal year 2024, the FDA charges $129,500 just to submit an original ANDA. If you need to make changes after approval-like switching suppliers or adjusting the manufacturing process-you’ll pay $5,000 per supplement. These are user fees, not taxes. They fund the FDA’s review process. Companies also spend $5-10 million on development, testing, and compliance. Compare that to the $2.3 billion it takes to launch a new brand drug.
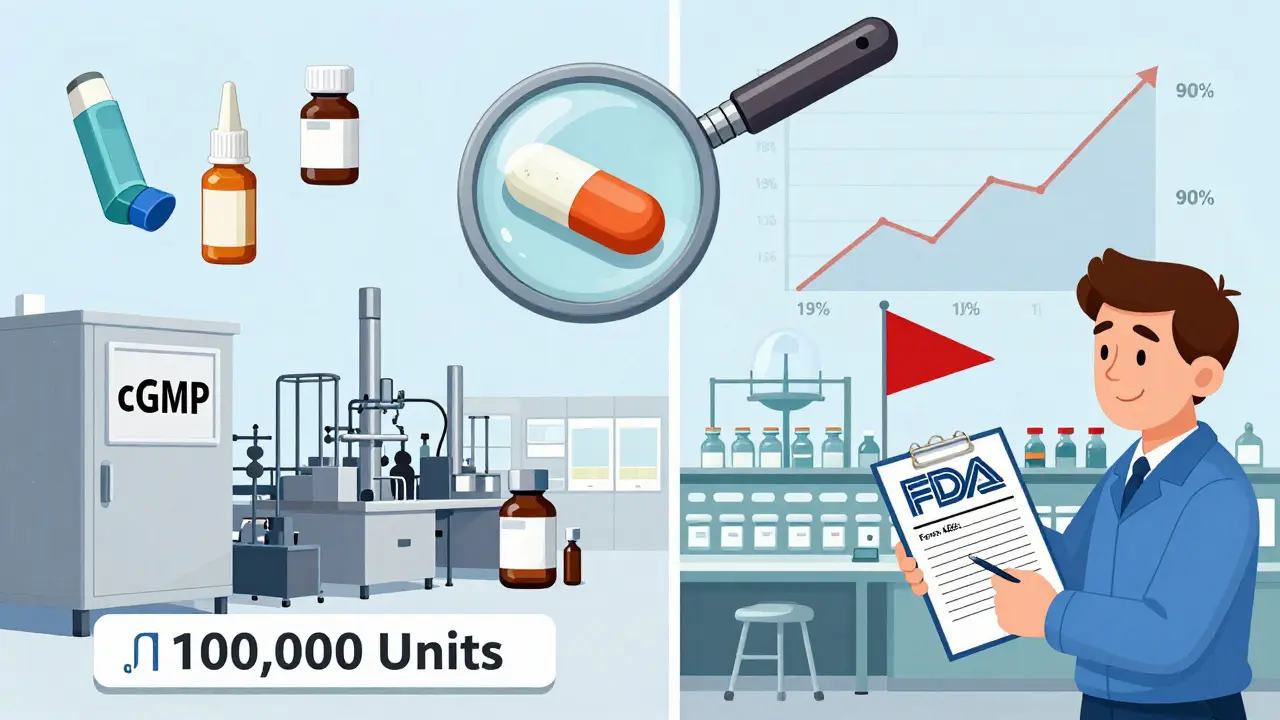
Manufacturing and Quality Rules
Generic drugs must be made under Current Good Manufacturing Practices (cGMP). The FDA inspects every facility-domestic or overseas-that produces the drug. In 2022, 68% of FDA Form 483 observations (which flag quality issues) came from foreign plants. The FDA requires exhibit batches: samples made at commercial scale (at least 10% of planned production size or 100,000 units, whichever is bigger). These are used to test stability and ensure the final product won’t break down before the expiration date. One common failure? Inadequate container closure validation. A single missed seal test can lead to a refusal to receive.
How ANDA Compares to Other Pathways
Not all generic applications are created equal. Here’s how ANDA stacks up:
| Pathway | Used For | Development Time | Cost | Clinical Data Required? |
|---|---|---|---|---|
| ANDA (505(j)) | Exact copy of brand drug | 3-5 years | $5-10 million | No |
| 505(b)(2) NDA | Modified version (new dose, route, etc.) | 7-9 years | $50-100 million | Partial |
| NDA (505(b)(1)) | New chemical entity | 10-15 years | $2.3 billion | Yes |
ANDA is for simple copies. If you want to change the drug in any meaningful way-like adding a new strength or changing the delivery system-you need a 505(b)(2) application. That’s more expensive and slower, but it’s the only legal way to make improvements on an existing drug without starting from scratch.
Common Reasons ANDAs Get Rejected
Over half of first-time ANDA submissions get deficiency letters. The most frequent reasons:
- Incomplete bioequivalence protocols: 28% of applications are refused because the study design doesn’t meet FDA standards. For example, using the wrong food state (fed vs. fasted) or too few subjects.
- Poor CMC documentation: 23% fail because manufacturing details are vague or lack validation. One company lost a $15 million project because they didn’t prove their tablet compression machine could consistently hit the right hardness.
- Patent certification errors: Misclassifying a patent as expired when it’s not can trigger a 30-month legal delay. One applicant waited 41 months because they filed a Paragraph IV certification incorrectly.
- Labeling mismatches: Even a small difference in warnings or dosing instructions can cause rejection.
Real-World Challenges
Companies report big differences in experience. Teva spent 42 months and $28 million trying to get a generic version of Advair Diskus approved, mainly because the inhaler device had to match the brand’s performance exactly. Meanwhile, Lupin got approval for a generic version of Jardiance in just 9.5 months by submitting a clean, complete application. The difference? Preparation. One company rushed. The other hired experienced regulatory staff, ran pre-ANDA meetings with the FDA, and triple-checked every document.

What’s Changing in 2025?
The FDA’s GDUFA III agreement (2023-2027) is pushing for faster reviews. By 2027, the goal is to approve 90% of standard ANDAs within 10 months and priority generics in 8 months. The agency is also investing $15 million in new tools to evaluate complex generics-like inhalers, nasal sprays, and topical creams-that are harder to copy. These products made up 35% of pending ANDAs in 2023, but only 42% were approved on first review. The FDA is also cracking down on “evergreening,” where brand companies file dozens of minor patents to block generics. The CREATES Act of 2019 already made it illegal to withhold samples needed for testing. New legislation in 2023 aims to limit patent thickets even further.
Who Can Navigate This Process?
It takes more than a good chemist. You need regulatory affairs professionals who understand FDA expectations, patent law, and quality systems. Most companies hire specialists with RAC (Regulatory Affairs Certified) credentials. These professionals earn an average of $125,000 a year. Training is expensive too-FDA workshops cost $450 per session. But the cost of getting it wrong? Millions. And years of delay.
Final Thoughts
The ANDA process isn’t easy, but it’s fair. It’s designed to ensure that a $5 generic pill works just as well as a $50 brand-name one. The legal requirements are strict because patient safety isn’t negotiable. For manufacturers, success comes from precision, patience, and preparation. For patients, it means access to affordable medicine. For the system, it means competition that works.
What happens if my ANDA gets refused to receive?
If the FDA refuses to receive your ANDA, it means your application is incomplete or doesn’t meet basic submission standards. You won’t get a review. You’ll get a letter listing the deficiencies-like missing forms, wrong format, or incomplete CMC data. You can resubmit after fixing the issues, but you’ll have to pay the fee again. Many companies use pre-ANDA meetings to avoid this.
Can a generic drug be approved if the brand drug is still under patent?
Yes, but only if the applicant files a Paragraph IV certification, claiming the patent is invalid or won’t be infringed. This triggers a 30-month legal stay, during which the FDA can’t approve the generic. The brand company can sue, and the court decides. If the patent is upheld, approval is delayed. If it’s invalidated, the generic can enter the market immediately.
How long does it take to get an ANDA approved?
Under GDUFA III targets, the FDA aims to approve 90% of standard ANDAs within 10 months of submission. But in practice, the average is 30-36 months due to deficiencies, inspections, and patent disputes. Complex products like inhalers or injectables often take longer-sometimes over 4 years.
Are generic drugs really as safe as brand-name drugs?
Yes. The FDA requires generics to meet the same strict standards for quality, strength, purity, and stability as brand drugs. The only difference is the cost. Over 90% of prescriptions filled in the U.S. are generics, and studies consistently show they perform the same in real-world use. Millions of patients rely on them every day.
Do I need to test my generic drug on humans?
You don’t need to test for safety or effectiveness again-that’s already proven by the brand drug. But you must run a bioequivalence study in healthy volunteers to prove your drug delivers the same amount of medicine into the bloodstream at the same rate. This is required by law for all ANDAs.
Next Steps for Manufacturers
If you’re considering entering the generic market, start here:
- Use the FDA’s Orange Book to find an off-patent brand drug with high sales volume and low complexity.
- Conduct a patent and exclusivity analysis-don’t assume a patent is expired.
- Reach out to the FDA for a pre-ANDA meeting to get feedback before spending millions.
- Hire experienced regulatory staff or consultants with proven ANDA success.
- Build or partner with a cGMP-compliant manufacturing facility.
- Invest in robust CMC documentation and bioequivalence study design.
The ANDA process is demanding, but it’s the most reliable way to bring affordable medicine to patients. Those who master it don’t just make money-they change lives.
Janette Martens
December 28, 2025 AT 17:11Marie-Pierre Gonzalez
December 29, 2025 AT 07:48Louis Paré
December 30, 2025 AT 02:46Samantha Hobbs
December 31, 2025 AT 15:14Nicole Beasley
January 1, 2026 AT 19:01sonam gupta
January 1, 2026 AT 19:52Julius Hader
January 3, 2026 AT 13:48Vu L
January 3, 2026 AT 23:16James Hilton
January 5, 2026 AT 00:31Mimi Bos
January 5, 2026 AT 06:01